Changes in regulatory requirements in both the United States and Europe, along with an increase in healthcare utilization, have significantly increased the number of MDR reports sent to the FDA. New innovation is a necessity if companies want to effectively handle the onslaught of complaints, investigations, and MDRs. Focusing on record collection and data entry alone is not sustainable, and new, modern methods should be developed.
The Increase in Volume of Complaints and MDR reports
In the past 10 years, the number of MDRs reported to the FDA has skyrocketed. In 2010, the number of submitted complaints was 337,838. In 2019, it had increased to 1,328,981. With the exception of 2016, the number of complaints filed increased by 20% each year. A conservative estimate shows that 1% of all complaints are MDRs, meaning that there are over 132 million complaints filed annually.
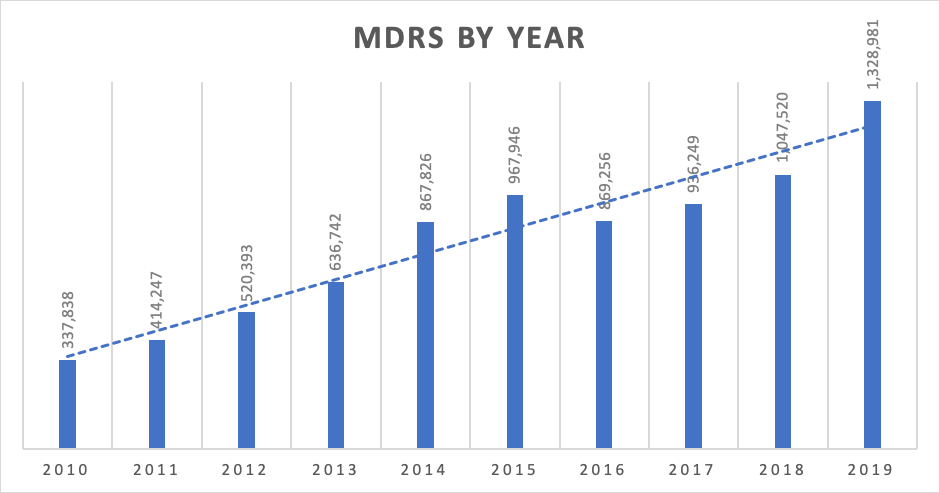
Picture 1 – MDRs by Year. 2010-2019
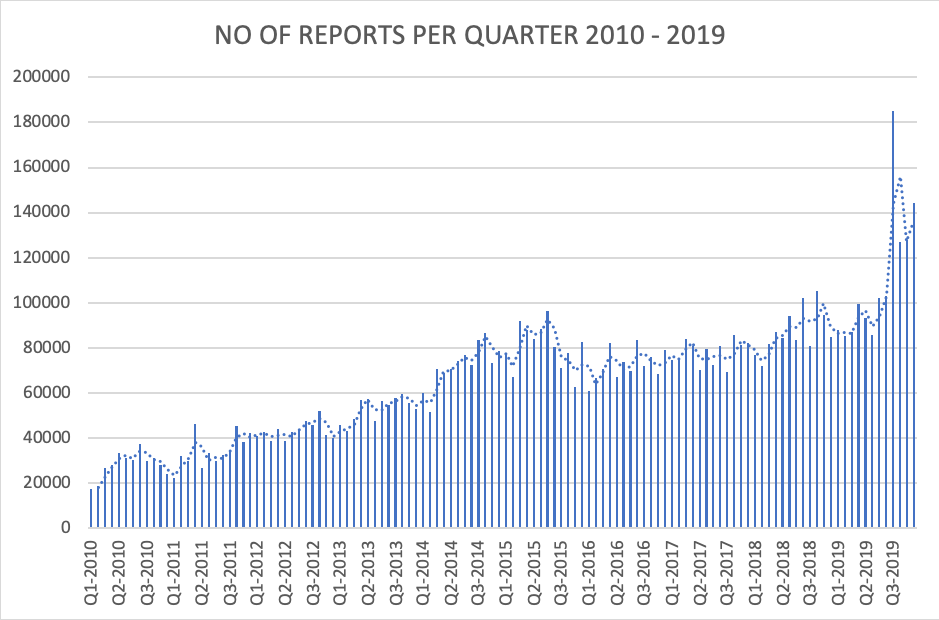
Picture 2 – MDRs by Quarter. 2010-2019
Compliance and Financial Costs of Post Market Surveillance
A high volume of complaints and MDRs have very pronounced implications for businesses. Handling complaints and MDRs is a complex process that must go through many voluminous stages necessary for their closure (Intake, Complaint Evaluations, Investigations, and Root Cause Analysis as well as Regulatory Reporting). The necessity of painstakingly reviewing MDRs and complaints means there is less time for companies to evaluate product performance, help R&D with new product development, and better communicate with Regulators.
Managing complaints on its own is not cheap; it costs the industry tens of billions of dollars per year. Depending on the device, the money a company may spend on a single MDR can be as high as $2000. These are unsustainable numbers, especially with the cost pressures of both government and private insurance.
Complaint Handling, MDRs and QMS
With the exception of a few very large Medical Device Manufacturers, Post Market Surveillance, Complaint Handling and Adverse Event Reporting/MDR have usually followed the lead of the Quality Departments by implementing Quality Management Software. That is because there is usually some connection with the Quality Teams in the area of CAPAs and Root Cause Investigations. When the volume of complaints and MDRs began its rapid expansion, it became evident that QMS would no longer be adequate. Post Market Surveillance is an extension of Customer Service more so than an extension of Quality Processes, which makes Complaint Handling and MDRs vastly more dynamic, prone to change and more difficult to maintain. The volume of field findings, product changes, regulatory updates and new product releases requires a different paradigm when it comes to software tools.
Post Market Surveillance Software and Automation
Medical Device Companies are no longer able to compromise in their operational processes in order to accommodate the software tools they have to use. The numbers are getting too large and companies are looking at a new generation of systems that are able to automatically respond to any future increase in Post Market Surveillance volume.
Medical Device Companies have stopped viewing Post Market Surveillance Software simply as a glorified spreadsheet. Collecting data and enforcing processes is not enough. The software tools needed must become the “extra person” that helps Medical Device Companies speed up complaint evaluation timelines, root cause investigations, and MDR reports. Automation is no longer a nice-to-have feature; It is absolutely necessary.

